A Toolbox Driven by Scientific Curiosity
Think of the Precision Health Initiative (PHI) as a toolbox. Only this toolbox holds
stem cells, sequencing platforms, cell models, and collaborations instead of hammers and
pliers. It’s built on the Rare Disease Discovery (RDD) Hub’s foundation, but it’s bigger,
bolder, and designed to do the deep dives that ordinary clinical pipelines can’t. Where RDD is
the genetic testing kit, PHI is the full workshop: precision immunology, cell modelling,
metabolite and protein analysis, and the people who know how to use them.
PHI’s purpose is simple and urgent: to take patients who’ve run out of clear answers in
standard clinical care and give them more ways forward. Instead of stopping at an exome or a
single genetic test, PHI layers on modelling technologies and lab-based experiments so
researchers can ask the next question: what does that variant actually do in a human cell?
That’s where the toolbox metaphor becomes literal: Put the patient’s variant into stem cells,
turn those into cardiac cells, watch how electrical currents behave, and you can study
arrhythmias, drug responses, and disease mechanisms outside the patient’s body. Isogenic
stem modelling (everything identical except one genetic change) lets us isolate cause and
effect without exposing patients to risk.
Practically, many of these experiments begin with a patient’s blood: peripheral blood cells are
reprogrammed into pluripotent stem cells (PSCs), which can then be differentiated into the
affected cell type. That workflow lets us compare a Variant of Uncertain Significance (VUS)
in a controlled way without needing tissue from the organ itself. For example,
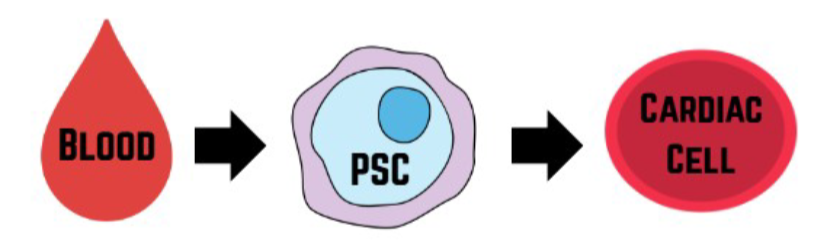
To edit the VUS out or replace it with the wild-type so the only difference between lines is
that single genetic change.
PHI doesn’t only work at the single-patient level. It scales to cohorts, builds representation,
and invests in population diversity because better science needs better samples. But
representation and modelling are expensive: exome tests, next-generation sequencing, and the
creation of stem cell lines all require significant funding; often thousands of dollars per
experiment. That’s why funding matters. Every tool added to this box expands what
researchers can test, model, and eventually translate back into care.
The team is working toward more accessible sequencing and on-site molecular profiling thus
increasing capacity for exome sequencing while also expanding in-person proteomics and metabolomics so clinicians can get richer, multidimensional readouts.
The Genome Library
Reading between the lines of DNA to find missing stories
Imagine the human genome to be an enormous library with twenty thousand books
placed on each shelf, each one telling a personalised story about how our bodies are built and
how they behave. On average, we’ve read around five thousand to eight thousand of these
books. They’re well-thumbed and memorised by heart. Yet some of these books are dusty and
written in barely recognisable dialect with missing footnotes. This is similar to our genes.
When a child has taken ill and the doctors are unable to provide answers, the Rare Disease
Discovery Hub saves the day like a gentle, brilliant librarian, detective team with curiosity,
patience, and a keen, third eye.
The standard genetic test that most doctors conduct is similar to a faithful magnifying
glass that solves a lot of mysteries. Yet often, the clues are tiny, hidden between pages or
folded into a margin note. And that’s when the Hub uses cutting edge technology similar to a
super microscope: Long-read sequencing! Think about a panoramic camera that reads entire
chapters in one go which is a lot more efficient that a singular person reading them. In
clinical terms, it’s a DNA sequencing method that reads tens and thousands of bases. The
second notable piece of technology used is RNA testing! This is similar to an advanced voice
recorder that listens to whether a gene’s message is being read correctly. These technologies
decipher genetic threads in the body coherently searching for hidden contradictions. Some of
these tests are research-only and aren’t yet part of routine clinical care that we explain clearly
from the beginning.
Every child we meet gets customised care: A large committee of clinicians,
researchers, and geneticists that observe the case and decide on the tools to be tried next. We
call in family witnesses as our secondary sources by testing parents and siblings to see
whether there’s a suspicious change shared by healthy relatives or if the illness only resides in
the affected child. This process is called segregation testing: a system similar to a Suspect
List when solving a mystery to separate the culprits from the innocent bystanders.
We’re honest.
This work while shrouded in technology has its limits. Sometimes tests
report a variant of uncertain significance: a genetic change we don’t yet know causes disease.
Of the twenty thousand human genes, scientists only clearly understand a few thousand well
enough to be able to tell you: “Yes, this gene abnormality causes that problem…”. Sometimes
we can be absolutely sure there’s a genetic answer from all our readings in this vast library
and still end up with a blank page of answers. We talk about this with families from the very
start. Genetic counsellors helpfully sit beside parents and patiently explain what tests might
or might not mean. There will never be any false hope for it is balanced with realism while
curiosity is balanced with intense care. We meet parents who’ve seen countless specialists
and our first job is to listen.
The speciality of the hub is how human it is in its mystery solving. Much like
Sherlock Holmes, we deal with rare cases across the province, our lab network and even the
world! We don’t solve mysteries alone but celebrate every small breakthrough because each
one teaches us about children everywhere and not just children in front of us. The work may
appear technical and slow but the simple most comforting part is always the same. Sitting with families, listening to their stories and doing every possible thing we can to transform the
“I don’t know” into “I believe I can see why”. This is a significant leap in science.
Think of the Rare Disease Discovery Hub as a warm, determined, ambitious library
where its detectives read between the lines to give you answers. Where translators adeptly
listen to the gene’s voices and where families are partners by giving us stories we won’t give
up searching.
Mapped Seas: The Hub’s Guide to Genetic Discovery
The research hub is like a mapped ocean where currents of clinical suspicion sweep
into a harbour, crates of samples are unloaded and a crew of technicians plot the seabed for
the genetic island that will finally give the answers to a child’s symptoms. It isn’t the regular
melodrama but a cartography with pipettes. Segregation cases are the charts. When a
clinician suspects a genetic cause, the Hub’s job is to trace whether that strange variant is an
inherited tide or a solitary wave. Families function as the compasses on the sailing sea.
Parents and patients alike are tested so that lab analysts can see if the same variant rides both
routes. If both patient and parent share the same variant, the current runs through the family.
If the variant appears only in the patient, that is a fresh island: A De novo change that has to
be explored from first principles.
The cargo arrives in many forms. Blood tubes are the sealed specimen crates that
come from scheduled clinic visits. Saliva arrives via mailed spit kits. Tiny bottled messages
that once returned are opened in the lab. Lysis buffer is delicately poured like a gentle tide;
breaking cells open and freeing DNA from its cellular moorings. The crude extract is washed
and precipitated like rinsing sand from a beach shell until the DNA is clean enough for
sequencing. Skin biopsies are treated differently: slivers of tissue are coaxed into fibroblast
cultures in T25 flasks, fed with specialised saline, and placed in incubators that hum a steady
climate: CO2, temperature, humidity, until the cultures grow into living archives. Those
cultures get frozen like heirloom seeds, stored for experiments that may come months or
years later.
Sequencing is the sonar. When the trace sings, the landscape is clear and when it’s
noisy everything starts to look like static. Technical tantrums are routine. A targeted PCR
refuses to amplify, a variant stubbornly hides or an enzyme underperforms. Troubleshooting
becomes an exercise in micro-adjustments, tweak the temperature a degree, swap reagents,
extend cycles until the signal sharpens. Families are generous partners, often sending fresh
samples without complaint. The bottleneck is usually a molecular quirk rather than human
patience. Discipline keeps the ship upright. Controls sit on separate benches and never mingle
with patient samples. A mislabelled tube would be a cartographic catastrophe. Every vial
bears a name and a barcode, every run has a checklist, and the lab’s rituals exist to prevent the
avoidable errors that wreck downstream interpretation.
Most cases resolve without the botanical detour of biopsies. Often blood is enough to
map inheritance. However, when deeper excavation is required, the Hub moves into Phase 2
territory which includes functional studies, RNA profiling, and experiments that probe how a
gene’s misbehaviour translates into biology. Those investigations are slower, less KPI-
friendly, and more exploratory which is the sort of work that generates maps valuable to
clinicians and funders alike. At the bottom, the Hub’s routine looks quite modest: add the
buffer, incubate, centrifuge, re-run. But the result matters. A clean sequence, a confirmed
inheritance pattern or a viable cell line is the difference between a family living in clinical
limbo and a clinician finally having a route forward. The lab’s day-to-day might read like
methodical tedium but its output is navigation. Precise coordinates handed back to families
and doctors so they can chart the next stretch of the journey